

-
Influence of the hydrogen-bond interactions on the excited-state dynamics of a push-pull azobenzene dye: the case of Methyl Orange
C. Nançoz, G. Licari, J.S. Beckwith, M. Soederberg, B. Dereka, A. Rosspeintner, O. Yushchenko, R. Letrun, S. Richert, B. Lang and E. Vauthey
Physical Chemistry Chemical Physics, 20 (10) (2018), p7254-7264


DOI:10.1039/C7CP08390D | Abstract | Article HTML | Article PDF | Supporting Info
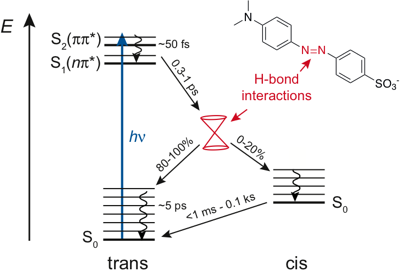
The excited-state dynamics of the pushâpull azobenzene Methyl Orange (MO) were investigated in several solvents and water/glycerol mixtures using a combination of ultrafast time-resolved fluorescence and transient absorption in both the UV-visible and the IR regions, as well as quantum chemical calculations. Optical excitation of MO in its trans form results in the population of the S2 ÏÏ* state and is followed by internal conversion to the S1 nÏ* state in â¼50 fs. The population of this state decays on the sub-picosecond timescale by both internal conversion to the trans ground state and isomerisation to the cis ground state. Finally, the cis form converts thermally to the trans form on a timescale ranging from less than 50 ms to several minutes. Significant differences depending on the hydrogen-bond donor strength of the solvents, quantified by the Kamlet Taft parameter α, were observed: compared to the other solvents, in highly protic solvents (α > 1), (i) the viscosity dependence of the S1 state lifetime is less pronounced, (ii) the S1 state lifetime is shorter by a factor of â1.5 for the same viscosity, (iii) the trans-to-cis photoisomerisation efficiency is smaller, and (iv) the thermal cis-to-trans isomerisation is faster by a factor of â¥103. These differences are explained in terms of hydrogen-bond interactions between the solvent and the azo nitrogen atoms of MO, which not only change the nature of the S1 state but also have an impact on the shape of ground- and excited-state potentials, and, thus, affect the deactivation pathways from the excited state.



